PSNtools for standalone and web-based structure network analyses of conformational ensembles
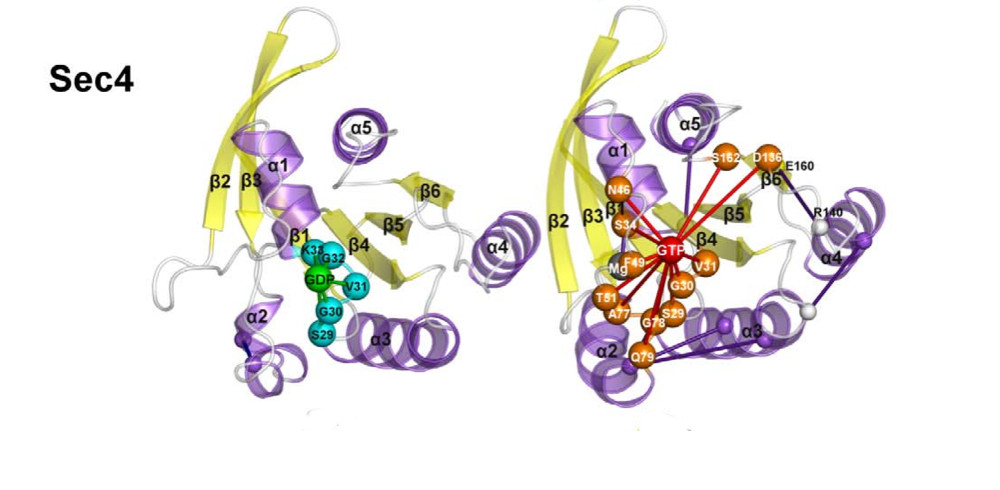
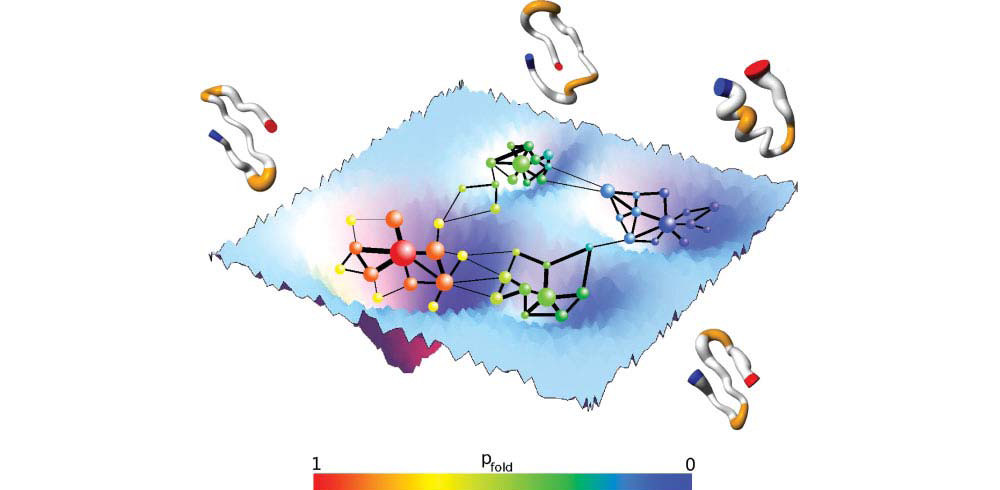
Welcome to the Computational Structural Biology Lab at the University of Modena and Reggio Emilia.


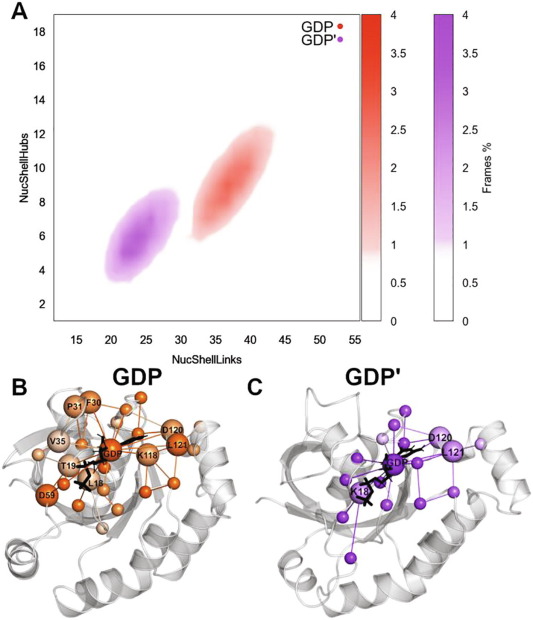
PSNtools for standalone and web-based structure network analyses of conformational ensembles
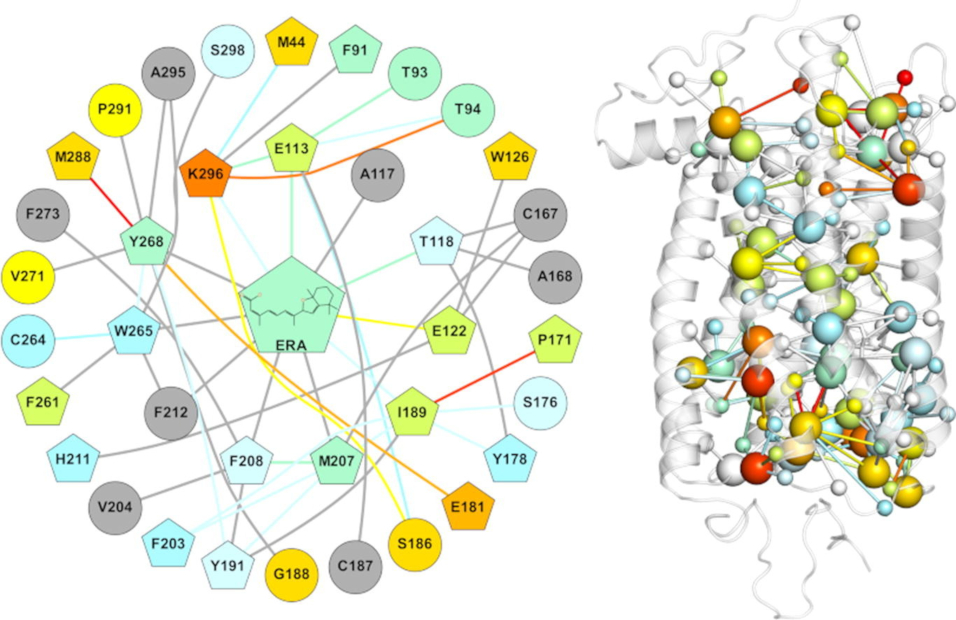
Structure network-based landscape of rhodopsin misfolding by mutations and algorithmic prediction of small chaperone action
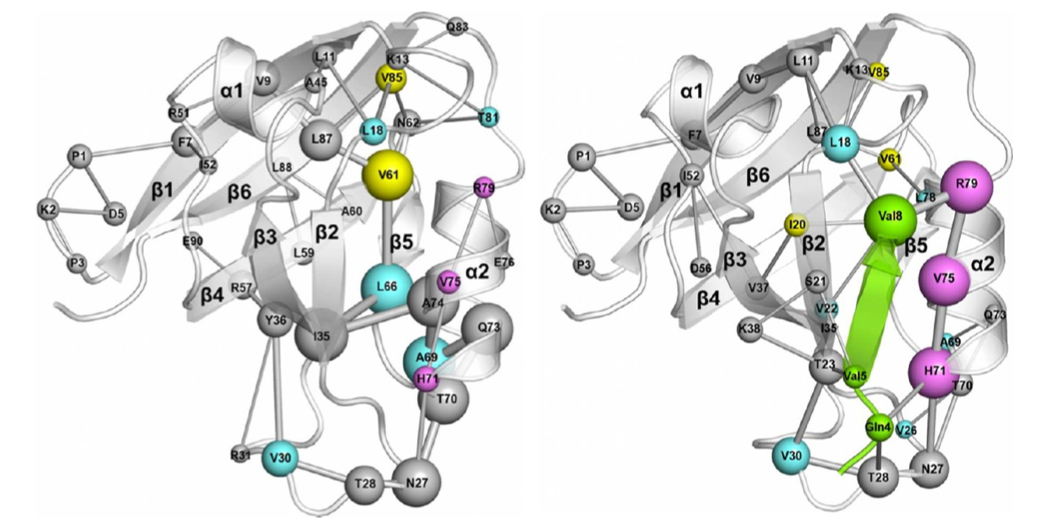
A Mixed Protein Structure Network and Elastic Network Model Approach to Predict the Structural Communication in Biomolecular Systems: The PDZ2 Domain from Tyrosine Phosphatase 1E As a Case Study
Welcome to the Computational Structural Biology Lab at the University of Modena and Reggio Emilia.